A região do MHC (Complexo Principal de Histocompatibilidade) está localizada no braço curto do cromossomo 6 e possui os genes que codificam as moléculas HLA (Antígeno Leucocitário Humano). O MHC é uma das regiões mais polimórficas do genoma humano e a genotipagem (ou tipificação) de indivíduos para os genes HLA é importante em muitas áreas da medicina e da pesquisa básica, tais como:
• Transplantes;
-medula óssea
-órgãos sólidos
-estudo de ligação com KIR
• Associação com doenças;
• Farmacogenética (resposta a medicamentos);
• Estudos de frequências populacionais;
• Antropologia e migrações humanas.
O transplante alogênico de medula óssea é o tratamento de escolha para muitas doenças hematológicas, tais como:
• Leucemia
• Aplasia medular
• Mieloma múltiplo
• Hemoglobinopatias
O sucesso do transplante de medula é dependente de vários fatores, incluindo o estágio da doença quando ocorre o transplante, o regime de condicionamento, a fonte de células tronco hematopoiéticas e o grau de identidade HLA entre doador e receptor.
A compatibilidade nos transplantes de medula é mais difícil, pois a análise é feita em ALTA RESOLUÇÃO, ou seja, é preciso identificar os dois alelos específicos do doador e do receptor, e não apenas os grupos alélicos, que é o que acontece em baixa resolução.
Porquê sequenciamento é o padrão ouro para tipagem HLA em alta resolução?
• A fronteira final da resolução
− Você visualiza seu alelo base por base.
• Resolução em 2, 4 ou até 6 dígitos na mesma reação
− Sequências acessórias com os GSSPs (máxima definição)
• Geração de dados por definitivo
− Resultados não são modificados por atualizações e desenhos de sondas.
• Fim dos problemas em relação a alelos raros
− Resultados não são baseados em similaridade de sequência (comum em SSP e SSO)
• Possibilidade de identificação de alelos nulos e novos alelos
Caso a mutação esteja presente nos éxons que você está analisando
A Invitrogen/Life Technologies comecializa os Kits SeCore®, que utilizam a química BigDye®, para o sequenciamento HLA. Estes kits oferecem as seguintes vantagens:
- Arquivos de dados analisados com software ABI;
- Sequenciadores Applied Biosystems já incluem calibração espacial e espectral para BigDye®;
- Maior resolução e qualidade.
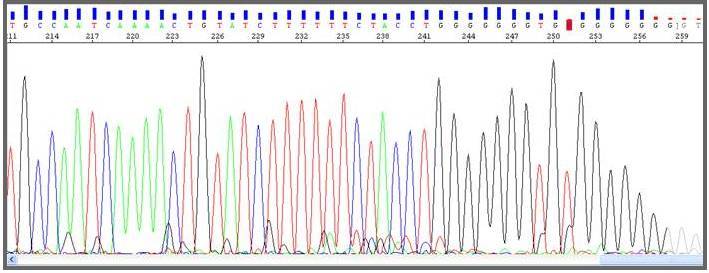
Fig. Genotipagem HLA com BigDye: melhor proporção de altura e distância entre os picos
Além disso, os kits Secore para tipagem HLA em alta resolução baseada em sequencia oferecem:
- Único programa de ciclagem para todos os loci, reduzindo erros de manuseio de amostras e melhorando a eficiencia.
- Amplificação curta, apenas 1,5 h – possibilitando amplificação e sequenciamento no mesmo dia.
- Identificação alélica robusta para todos os loci e picos de alelos balanceados para identificação de heterozigozidade.
- Ampla lista de kits GSSP (Group Specific Sequencing Primers) para resolução de ambiguidades.
A estratégia Secore começa com a amplificação do locus alvo através de mix de amplificação, Taq DNA Polimerase Fast Start e amostra de DNA genomico. O produto resultante é tratado com ExoSAP-IT antes do sequenciamento, para degradar primers não incorporados e hidrolizar nucleotídeos livres. A sequencia de nucleotideos e o subtipo HLA resultante é determinado por sequenciamento multicolorido baseado em fluorescência. As reações finais são purificadas através de uma precipitação com etanol antes da leitura. As amostras desnaturadas são carregadas e os resultados detectados em um instrumento de sequenciamento automatizado.
Cada kit Secore inclui:
- Primers loci especificos
- Taq DNA Polimerase Fast Start
- Enzima ExoSAP-IT
- Mix de sequenciamento (primers, corantes, terminadores e polimerase)
- Tampão de precipitação
O software U-Type
É o software utilizado para a análise dos dados. O Software faz o base calling das amostras, montagem da sequencia consenso e identificação contra uma biblioteca das sequências dos alelos descritos de cada loco.
O Software U-Type também sugere kits GSSP que podem ser usados na resolução de ambiguidades, quando essas são observadas. Estes kits utilizam a técnica de SSP para resolver situações em que dois ou mais gonótipos igualmente prováveis são sugeridos para o mesmo indivíduo.